系统发育研究是利用形态、生理生化或分子等数据推断物种之间的进化关系。现在用的最多的是利用蛋白质或核酸序列推断物种的系统发育树(也叫系统发生树),这种系统发育树称为分子系统发育树。
在构建分子系统发育树前,需要先对序列进行多序列比对,比对的工具有Clustal(包括图形界面的ClustalX和命令行界面的ClustalW,以及Clustal Omega等)、MUSCLE、T-COFFEE等。
构建系统发育树的方法也有很多种,如距离法、最大似然法、最大简约法、贝叶斯方法等,软件有PHYLIP、MEGA、PAUP、PAML、PHYML、FastTree、MrBayes等。
从序列开始构建系统发育树的步骤比较繁琐,我们可以把这些步骤写成shell脚本,通过循环就可以实现批量构建系统发育树,这个在Windows下用图形界面工具是很难实现的。
文件lde.fa中是7个物种的套索RNA去分支酶(lariat debranching enzyme)蛋白的序列,这种蛋白质在这些物种中只有一个拷贝,保证相互之间是直系同源关系,从而推断出的基因树能代表物种树。下面只显示了前两个物种小鼠和人的序列:
[xiezy@ibi98 ml]$ head -22 lde.fa
>Musmu ENSMUSP00000070991.5 protein_coding gene_symbol:Dbr1 description:debranching
RNA lariats 1
MRVAVAGCCHGELDKIYETLALAERRGSGPVDLLLCCGDFQAVRNEADLRCMAVPPKYRH
MQTFYRYYSGEKKAPVLTIFIGGNHEASNHLQELPYGGWVAPNIYYLGLAGVVKYRGVRI
GGISGIFKSHDYRKGHFECPPYNSSTIRSIYHVRNIEVYKLKQLKQPVHIFLSHDWPRNI
YHYGNKKQLLKTKSFFRQEVENSTLGSPAASELLEHLQPAYWFSAHLHVKFAALMQHQAT
DKDQAGKETKFLALDKCLPHRDFLQVLEIEHDPSAPEYLEYDVEWLTVLRATDDLINVTG
GLWNMPEDNGLHTRWDYSATEETMKEVMEKLNHDPKVPCNFTMTAACYDPSKPQTQVKLV
HRINPQTTEFCAQLGITDINVMIQKAREEEHHQCGEYEQQGDPGTEESEEDRSEYNTDTS
ALSSINPDEIMLDEEEEEEEEEEEAVSAHSDMNTPSVEPASDQASDLSTSFSDIRNLPSS
MFVSSDDASRSPASGEGKCGETVESGDEKDLAKFPLKRLSDEHEPEQRKKIKRRNQAIYA
AVDDGDASAE
>Homsa ENSP00000260803.4 protein_coding gene_symbol:DBR1 description:debranching
RNA lariats 1
MRVAVAGCCHGELDKIYETLALAERRGPGPVDLLLCCGDFQAVRNEADLRCMAVPPKYRH
MQTFYRYYSGEKKAPVLTLFIGGNHEASNHLQELPYGGWVAPNIYYLGLAGVVKYRGVRI
GGISGIFKSHDYRKGHFECPPYNSSTIRSIYHVRNIEVYKLKQLKQPIDIFLSHDWPRSI
YHYGNKKQLLKTKSFFRQEVENNTLGSPAASELLEHLKPTYWFSAHLHVKFAALMQHQAK
DKGQTARATKFLALDKCLPHRDFLQILEIEHDPSAPDYLEYDIEWLTILRATDDLINVTG
RLWNMPENNGLHARWDYSATEEGMKEVLEKLNHDLKVPCNFSVTAACYDPSKPQTQMQLI
HRINPQTTEFCAQLGIIDINVRLQKSKEEHHVCGEYEEQDDVESNDSGEDQSEYNTDTSA
LSSINPDEIMLDEEEDEDSIVSAHSGMNTPSVEPSDQASEFSASFSDVRILPGSMIVSSD
DTVDSTIDREGKPGGTVESGNGEDLTKVPLKRLSDEHEPEQRKKIKRRNQAIYAAVDDDD
DDAA
下面是从Fasta格式的序列开始利用FastTree构建系统发育树的shell脚本fasta_ml_tree.sh:
[xiezy@ibi98 ml]$ cat fasta_ml_tree.sh
#!/bin/bash
###############################################################
# This shell used to make ML tree using ClustalW and FastTree #
# from Fasta protein file. Written by Zengyan Xie. #
###############################################################
if [ $# != 1 ]
then
echo "Usage: $0 ProteinFile(Fasta format)"
exit 1
fi
infile=$1
basename=`echo $infile |cut -d'.' -f1`
# alignment
echo "Alignning ......"
clustalw2 <<eof >/dev/null
1
$infile
2
9
4
1
x
x
eof
# build ML tree using FastTree
echo "Building ML tree (FastTree) ......"
FastTree $basename.phy >$basename.ML.tre 2>/dev/null
echo "Done. Phylogenetic tree is saved to file $basename.ML.tre"
clustalw程序在运行后需要根据提示输入参数,我们把这些参数利用即时输入重定向提供给程序(空行是一次回车),这样就不需要在程序运行时再输入参数。另外,clustalw和FastTree在运行时会有一些提示,我们把这些提示重定向到/dev/null(没有输出),并输出正在运行的步骤的提示。
下面是用该脚本从上面的蛋白质序列推断物种关系,最后得到Newick格式的系统发育树文件:
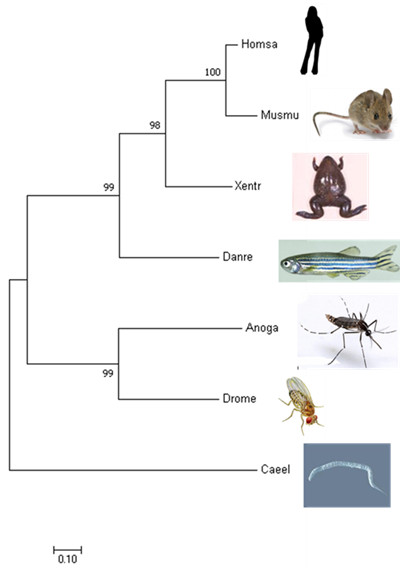
[xiezy@ibi98 ml]$ ./fasta_ml_tree.sh lde.fa Alignning ...... Building ML tree (FastTree) ...... Done. Phylogenetic tree is saved to file lde.ML.tre [xiezy@ibi98 ml]$ cat lde.ML.tre (Xentr:0.23934,(Musmu:0.11180,Homsa:0.04192)1.000:0.21813,(Danre:0.36023,(Caeel:0.95772, (Anoga:0.44563,Drome:0.36099)0.996:0.32881)0.998:0.33029)0.989:0.16692);
将该文件下载到本地,利用MEGA或TreeView可以查看(图中的图片为另外添加),图中节点上的数字为检验值百分比,该值越高表示该节点下的分支聚在一起的可信度越高。
本例利用shell脚本,将多序列比对和系统发育树构建的软件进行封装,一个命令就可以从Fasta格式的序列文件,生成系统发育树,并可以反复使用或通过循环一次构建大量的系统发育树。